高斯玻色采样模拟分子振动光谱#
此研究涉及两个主要领域。其中之一是关于量子信息和量子计算机的研究。我们选择玻色采样进行讨论。 目前,有多种物理实现方式可以进行玻色子取样,但最常见的是光子解决方案。 在该方案中,单光子态被用作输入到干涉仪,在输出端测量光子数。 干涉装置相对简单;而制备单光子和测量光子数对仪器的要求较高。 因此,如果我们考虑使用相干激光,可以让实验简便一些。 由于相干态可以用高斯分布描述,这种类型的玻色子采样被称为高斯玻色采样。
另一个主要领域是光谱学。我们以分子振动光谱为例,模拟不同带电态之间转变的概率,从而近似确定分子振动谱。 这种方法可以使用高斯玻色采样来实现。我们在基于高斯玻色采样的模拟器上实现该模拟过程。
高斯玻色采样#
量子比特是一个抽象的二维量子物理系统。 其希尔伯特空间通常可由两个基态 \(\left|0\right\rangle\) 和 \(\left|1\right\rangle\) 来描述。 不同量子比特可以处于叠加态和纠缠状态。 我们可将其推广到3维、4维以至有限的 \(d\)-维空间中。
以上内容可以进一步推广到无限维度。 玻色子粒子是这种类型的量子物理系统的一个例子,其中在给定状态中可以有无限多个粒子。 这样的系统仍然是量子系统,仍具有叠加和纠缠性。 在可数无限维度情况下,量子态被称为 qumodes。
在真实物理系统中,任何物理量都需要通过多次测量来确定其状态分布。 因此量子计算机都是对抽样问题的物理实现。 在可数无限维度的情况下,这被称为玻色子采样。
在多种玻色采样的物理实现方式中,最简单的仍然是光子方案。 量子光学设备在 qumodes 上为幺正算符。常用高斯算符如下:
事实上,一个纯的光子数态,即 \(\left|n\right\rangle\), 在技术上很难做到。 \(\left|1\right\rangle\) 意味着特定量子模式中只有一个光子。 精确地产生一个光子是一项巨大挑战。 而相干光是每个量子模式的可数无限状态之和,高斯玻色采样以相干光作为输入,是一种更简单的解决方案。
分子的振动光谱简要描述#
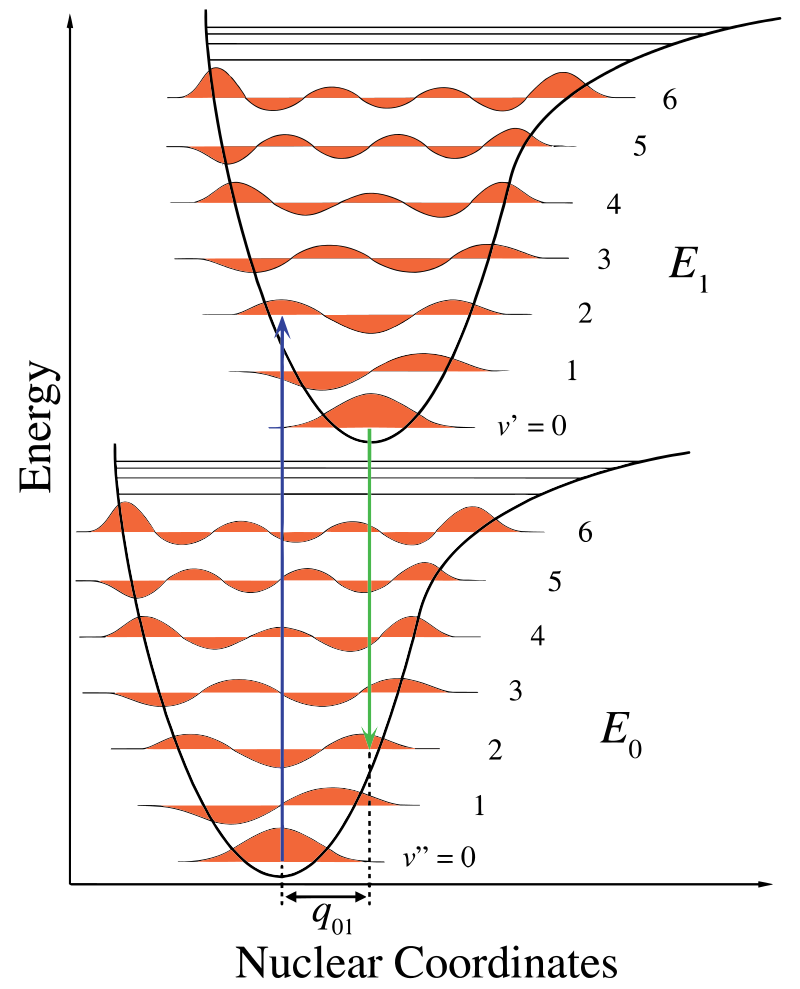
光谱学的一个主要问题是检查原子和分子之间电荷状态的跃迁。 分子吸收的光频率便是取决于不同电子态之间的允许跃迁。 这些电子跃迁可能伴随着分子振动能量的变化。 在这种情况下,代表更强烈吸收光的频率的吸收线被称为振动光谱。 然而,除了氢原子可以被精确解决外,随着电子数量增加,量子力学方程变得指数级复杂,使得对它们的计算变得不可能。因此必须使用近似方法。
如果假设核和电子状态是独立的,对于极其复杂的分子系统,一个很好的近似称为玻恩-奥本海默近似。 由于原子核的质量较大,其对电子壳层变化反应更慢,因此在 Franck-Condon 原理中可以将原子核视为恒定。
通过用简单的量子谐振子来近似电子的振动,可以将电子的哈密顿算符写成反应坐标的函数。 然后,电子势能表面已经可以用抛物线描述。
Duschinsky 提出一种近似方法,不同电子态的简正坐标之间存在线性关系
Doktorov 证明,在这些条件下,对于这样的量子谐振子,不同势能面上的量子态 \(\left|\phi\right\rangle\) 和 \(\left|\phi'\right\rangle\) 之间的关系可以用以下方式给出:
其中,Doktorov 算子定义为
旋转算符的输入参数是Duschinsky混合矩阵本身
位移算符D的参数是Duschinsky位移
压缩算符是从分子的物理特性推导出来的。\(\Omega\) 和 \(\Omega'\) 是分子内原子在发生跃迁前后谐波的角频率
上面哈密顿量的本征值问题的解是 Fock 空间中的相干态。高斯算符作用于它们之后,将一个相干态转换为另一个相干态。 不同电子态之间的转移概率,即所谓的 Franck-Condon 因子,可以近似表示为
玻色子采样的计算在于对 qumodes 进行旋转矩阵操作。 在我们计算振动光谱时,使用上面的 Doktorov 算符。其过程为
制备相干态
将相干态传输到一个玻色采样设备,以获得转移概率
进行第二次压缩,测量各模式分布,进一步计算FCF
Hessian matrix#
对于一双原子分子,假设谐振子势能
其中 \(r\) 是两个核之间的距离,\(r_e\) 是它们的平衡距离。 简单起见,可只考虑X轴。我们可以用两个原子的笛卡尔坐标 \(x_1\)(对应原子1)和 \(x_2\)(对应原子2)来表示这一点。 则,\(r = x_2 - x_1\)。
势能的二阶导数是:
Hessian矩阵(仅针对x方向)为:
质量加权的Hessian矩阵是:
如果我们有了系统的Hessian矩阵和质量加权Hessian矩阵,就可以由特征向量定义质量加权简正模式。 接着,利用分子的平衡结构坐标数据、质量加权简正坐标等数据可以求解所需高斯算符参数。
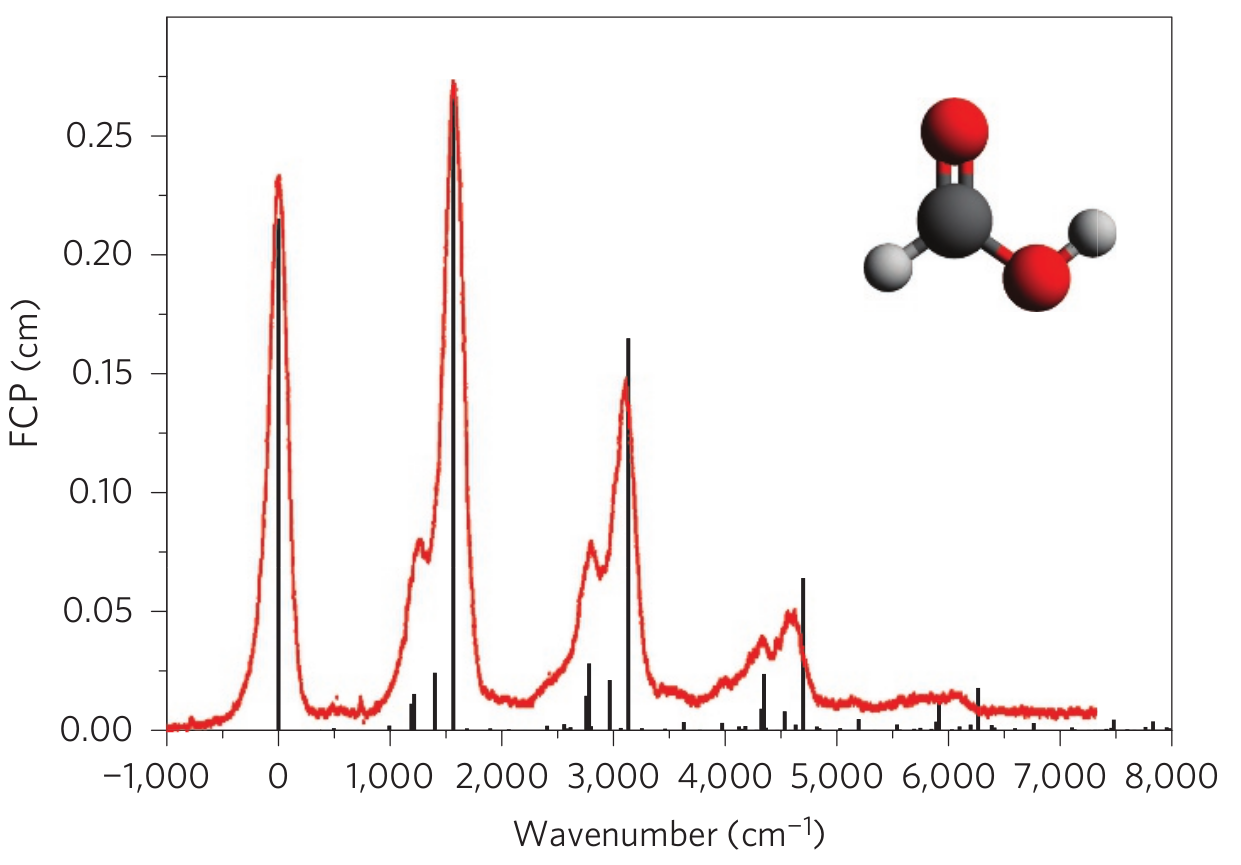
Jankowiak的补充材料提供了一些分子的相关数据。 我们以甲酸(\(1^{1}A'\rightarrow1^{2}A'\))为例计算分子振动光谱:
import deepquantum as dq
import matplotlib.pyplot as plt
import numpy as np
import torch
\(1^1A'\) 态及\(1^2A'\) 态的平衡结构坐标如下
ri = np.genfromtxt('./data/formic_ri.csv', delimiter=',', skip_header=0)[:, np.newaxis]
rf = np.genfromtxt('./data/formic_rf.csv', delimiter=',', skip_header=0)[:, np.newaxis]
\(1^1A'\) 态及 \(1^2A'\) 态的质量加权简正坐标如下
li = np.genfromtxt('./data/formic_li.csv', delimiter=',', skip_header=0)
lf = np.genfromtxt('./data/formic_lf.csv', delimiter=',', skip_header=0)
\(1^1A'\) 态及 \(1^2A'\) 态简正模式频率如下
omega = np.genfromtxt('./data/formic_omega.csv', delimiter=',', skip_header=0)
omegap = np.genfromtxt('./data/formic_omegap.csv', delimiter=',', skip_header=0)
计算中用到的物理学常量
c = 299792458.0 # 光速
mu = 1.6605390666 * 10**-27 # 原子质量单位
h = 6.62607015 * 10**-34 # 普朗克常数
m_c = 12 # 碳原子相对原子质量
m_h = 1.007825037 # 氢原子相对原子质量
m_o = 15.994914640 # 氧原子相对原子质量
Duschinsky 矩阵 \(U\) 及位移矢量 \(\delta\) 计算如下
u = []
for li_ele in li:
for lf_elf in lf:
u.append(np.sum(li_ele * lf_elf))
u = np.array(u[-1::-1]).reshape(7, 7).T
print(u)
array([[ 0.99343181, 0.01440011, 0.01532633, 0.02861045, 0.06378083,
0.07513988, -0.04280796],
[-0.01485231, 0.99314303, 0.07419536, 0.0769291 , -0.03610189,
-0.00248855, 0.01727882],
[-0.01185718, -0.09164895, 0.84227531, 0.17994129, -0.38567948,
0.30738928, 0.08008576],
[ 0.03813492, 0.04087165, -0.34031972, -0.52311845, -0.66785609,
0.38477092, 0.11420873],
[-0.04133251, -0.03419326, -0.40036477, 0.76362651, -0.10356638,
0.48381836, 0.09406589],
[ 0.0907918 , -0.04184572, -0.09066643, 0.31511355, -0.59003401,
-0.719268 , 0.13037051],
[-0.03245558, 0.00500992, -0.02063661, 0.06935002, -0.20181377,
0.01732396, -0.97588364]])
delta = []
m = np.diag([m_c, m_c, m_c, m_o, m_o, m_o, m_o, m_o, m_o, m_h, m_h, m_h, m_h, m_h, m_h])
for i in range(len(omegap)):
d = lf[i].T @ np.sqrt(m) @ (ri - rf)
denominator = np.sqrt(h / (4 * np.pi**2 * 100 * omegap[i] * c * mu)) / (10**-10)
delta.append(d / denominator)
delta = np.array(delta[-1::-1])
print(delta)
array([[ 0.22536617],
[ 0.14689208],
[ 1.55989779],
[-0.37838396],
[ 0.45525871],
[-0.34391138],
[ 0.06184607]])
在用于计算振动光谱的GBS算法中,以上这些化学参数足以确定GBS设备的配置。 利用这些数据,我们即可计算甲酸分子振动光谱。
实际上,在可能仅涉及有限数量的光子的情况下需要非线性相互作用,第二次挤压操作在光学装置中难以实现。 通常,我们需要将两次挤压操作压缩为一次:
其中 $\(J=\Omega'U\Omega^{-1}\)\( \)\(\delta=\hbar^{-1/2}\Omega'd\)\( \)\(J=C_{L}\Sigma C_{R}^{t}\)$
各 GBS 参数计算如下:
pre_transition_squeezing = np.sqrt(omega[-1::-1])
post_transition_squeezing = np.sqrt(omegap[-1::-1])
j_mat = np.diag(post_transition_squeezing) @ u @ np.linalg.inv(np.diag(pre_transition_squeezing))
cl, lambda_1, cr = np.linalg.svd(j_mat)
delta_2 = np.linalg.inv(j_mat) @ delta / np.sqrt(2)
delta_2 = delta_2.flatten()
lambda_2 = np.log(lambda_1)
甲酸分子振动光谱计算如下所示#
modes = 7 # 简正模式数量
cutoff = 3
shots = 500000
cir = dq.photonic.QumodeCircuit(
nmode=modes,
init_state='vac',
# init_state=init_state,
cutoff=cutoff,
backend='gaussian',
)
for i in range(modes):
cir.d(wires=[i], r=delta_2[i])
cir.any(cr, wires=list(range(modes)))
for i in range(modes):
cir.s(wires=[i], r=-lambda_2[i])
cir.any(cl, wires=list(range(modes)))
state = cir()
# 线路可视化
cir.draw()

sample = cir.measure(shots=shots, mcmc=True)
Using MCMC method to sample the final states!
chain 1: 0%| | 0/99999 [00:00<?, ?it/s]
chain 1: 100%|██████████████████████████| 99999/99999 [00:22<00:00, 4435.32it/s]
chain 2: 100%|█████████████████████████| 99999/99999 [00:01<00:00, 90359.99it/s]
chain 3: 100%|█████████████████████████| 99999/99999 [00:01<00:00, 90929.04it/s]
chain 4: 100%|█████████████████████████| 99999/99999 [00:01<00:00, 91488.54it/s]
chain 5: 100%|█████████████████████████| 99999/99999 [00:01<00:00, 93091.03it/s]
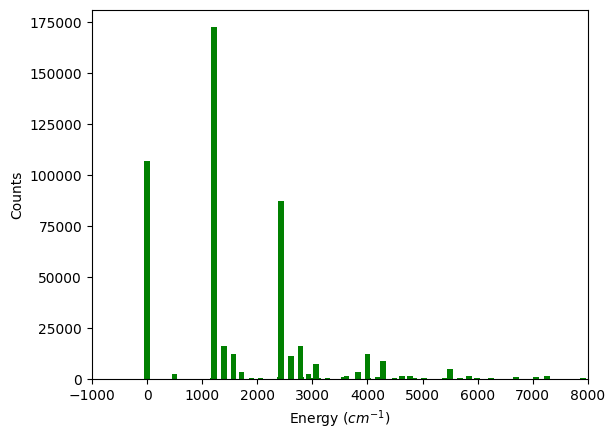
wave_number = []
counts = []
for ele in sample.items():
# print(ele[0].state)
wave_number.append(torch.sum(ele[0].state * torch.tensor(omegap)))
counts.append(ele[1])
plt.bar(wave_number, counts, width=100, color='g')
plt.xlabel(r'Energy $(cm^{-1})$')
plt.ylabel(r'Counts')
plt.xlim(-1000, 8000)
plt.show()
参考结果#